Eric Almeida, Kevin Luczak, Dasith Perera
RNA Sequencing is used to investigate the transcriptional profile of a cell
Genome (DNA) -> Transcriptome (RNA) -> Protein
RNA Transcripts are used to:
- transcribe proteins
- form the ribosomal translation machinery
- regulate gene expression
- modulate proteins
RNA can be sequenced using Next Generation Sequencing

? 2017 Illumina, Inc. All rights reserved.
Illumina sequencing-by-synthesis
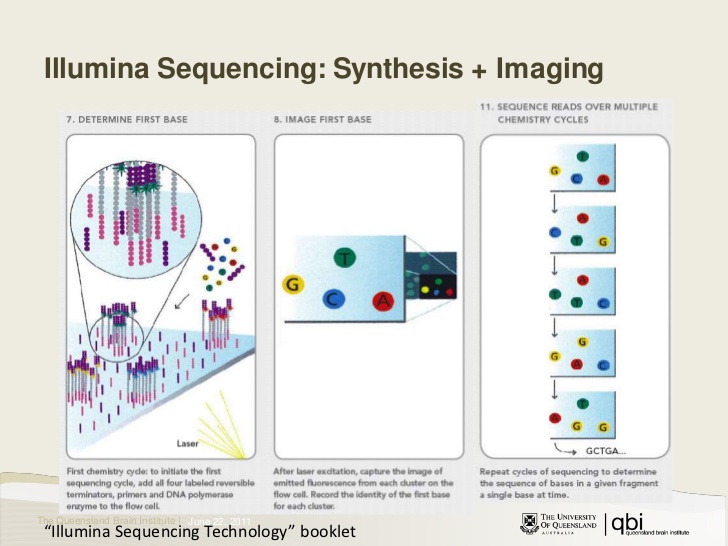
Example RNA-sequencing workflow
- Step 1. Experimental Design
- Step 2. Library Preparation
- Step 3. Execute the experiment
- Step 4. Align sequencing data to reference genome using BowTie2
- Step 5. Obtain count data using htseq-count
- Step 6. Move data into R and generate results
NOISeq Install from Bioconductor
source("https://bioconductor.org/biocLite.R") #Downloading from Bioconductor
biocLite("NOISeq")
library(NOISeq)
Help Viewer in R
Documentation and help can be easily accessed through the “? (command)” command
In example:
-
?head # takes you to Help viewer for ‘head’ command
-
?noiseq # takes you to Help viewer for noiseq package
View Input Data
Load your data into R and make your own treatment groups
data(Marioni)
head(mycounts) #Count data (data from upstream)
## R1L1Kidney R1L2Liver R1L3Kidney R1L4Liver R1L6Liver
## ENSG00000177757 2 1 0 0 1
## ENSG00000187634 49 27 43 34 23
## ENSG00000188976 73 34 77 56 45
## ENSG00000187961 15 8 15 13 11
## ENSG00000187583 1 0 1 1 0
## ENSG00000187642 4 0 5 0 2
## R1L7Kidney R1L8Liver R2L2Kidney R2L3Liver R2L6Kidney
## ENSG00000177757 2 0 1 1 3
## ENSG00000187634 41 35 42 25 47
## ENSG00000188976 68 55 70 42 82
## ENSG00000187961 13 12 12 20 15
## ENSG00000187583 3 0 0 2 3
## ENSG00000187642 12 1 9 4 9
head(myfactors) #Treatment groups / replicates as you define it
## Tissue TissueRun
## R1L1Kidney Kidney Kidney_1
## R1L2Liver Liver Liver_1
## R1L3Kidney Kidney Kidney_1
## R1L4Liver Liver Liver_1
## R1L6Liver Liver Liver_1
## R1L7Kidney Kidney Kidney_1
myfactors = data.frame(Tissue = c("Kidney", "Liver", "Kidney", "Liver", "Liver", "Kidney", "Liver", "Kidney", "Liver", "Kidney"), TissueRun = c("Kidney_1", "Liver_1", "Kidney_1", "Liver_1", "Liver_1", "Kidney_1", "Liver_1", "Kidney_2", "Liver_2", "Kidney_2"), Run = c(rep("R1", 7), rep("R2", 3)))
myfactors #Should see the other defined treatment groups
## Tissue TissueRun Run
## 1 Kidney Kidney_1 R1
## 2 Liver Liver_1 R1
## 3 Kidney Kidney_1 R1
## 4 Liver Liver_1 R1
## 5 Liver Liver_1 R1
## 6 Kidney Kidney_1 R1
## 7 Liver Liver_1 R1
## 8 Kidney Kidney_2 R2
## 9 Liver Liver_2 R2
## 10 Kidney Kidney_2 R2
Additional Bioinformation, from Ensembl Biomart
Provided for us in the Marioni data set, but easy to DIY. In example, first download the unique gene ID and biotype to a csv file from an online database that contains this information on your organism of study, then:
#GENES <- read.csv("C:/Users/labuser/Documents/R/GENES.txt")
#GENES1 <- data.frame(GENES$'Gene.stable.ID')
#GENES1$Gene.Type <- GENES$'Gene.type'
#GENES2 <- unique(GENES1)
#ID <- GENES2$'GENES.Gene.stable.ID'
#TYPE <- GENES2$'Gene.Type'
#BIOTYPES <- data.frame(TYPE)
#row.names(BIOTYPES) <- GENES2$'GENES.Gene.stable.ID'
Creating a NOISeq Object
mydata <- readData(data = mycounts, length = mylength, gc = mygc, biotype = mybiotypes, chromosome = mychroms, factors = myfactors)
mydata
## ExpressionSet (storageMode: lockedEnvironment)
## assayData: 5088 features, 10 samples
## element names: exprs
## protocolData: none
## phenoData
## sampleNames: R1L1Kidney R1L2Liver ... R2L6Kidney (10 total)
## varLabels: Tissue TissueRun Run
## varMetadata: labelDescription
## featureData
## featureNames: ENSG00000177757 ENSG00000187634 ...
## ENSG00000201145 (5088 total)
## fvarLabels: Length GC ... GeneEnd (6 total)
## fvarMetadata: labelDescription
## experimentData: use 'experimentData(object)'
## Annotation:
QC Plots
Biases (error) are introduced through RNA extraction, library preparation, and sequencing processes
Quality control tests offered by NOISeq:
- Biotype detection
- QC Bias
- Length Bias
- RNA composition
Biotype Detection
- Used to quickly visualize potential contamination or technical biases introduced during library preparation
mybiodetection <- dat(mydata, k=0, type = "biodetection", factor = NULL)
## Biotypes detection is to be computed for:
## [1] "R1L1Kidney" "R1L2Liver" "R1L3Kidney" "R1L4Liver" "R1L6Liver"
## [6] "R1L7Kidney" "R1L8Liver" "R2L2Kidney" "R2L3Liver" "R2L6Kidney"
par(mfrow = c(1,2))
explo.plot(mybiodetection, samples = c(1,2), plottype ="persample")
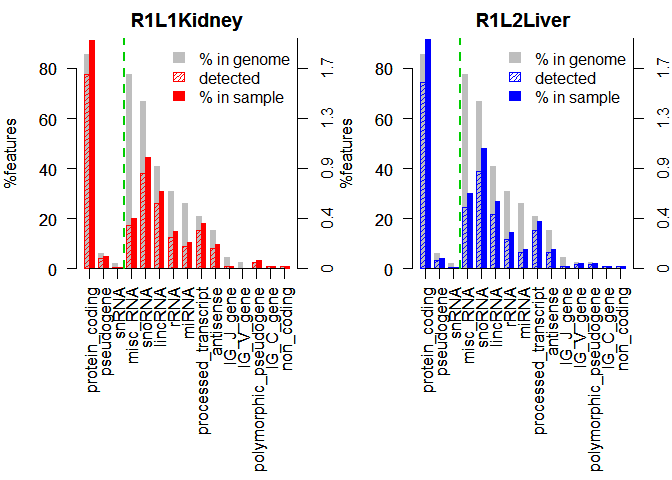
Biotype Detection, output 2
mybiodetection <- dat(mydata, k=0, type = "biodetection", factor = NULL)
## Biotypes detection is to be computed for:
## [1] "R1L1Kidney" "R1L2Liver" "R1L3Kidney" "R1L4Liver" "R1L6Liver"
## [6] "R1L7Kidney" "R1L8Liver" "R2L2Kidney" "R2L3Liver" "R2L6Kidney"
par(mfrow = c(1,2))
explo.plot(mybiodetection, samples = c(1,2), toplot = "protein_coding", plottype ="comparison")
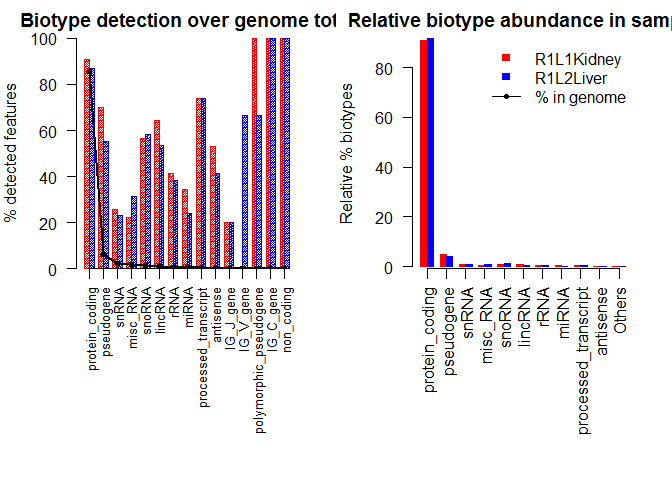
## [1] "Percentage of protein_coding biotype in each sample:"
## R1L1Kidney R1L2Liver
## 91.0181 91.7060
## [1] "Confidence interval at 95% for the difference of percentages: R1L1Kidney - R1L2Liver"
## [1] -1.7991 0.4233
## [1] "The percentage of this biotype is NOT significantly different for these two samples (p-value = 0.2302 )."
Biotype Detection, output 3
mycountsbio <- dat(mydata, factor = NULL, type = "countsbio")
## [1] "Count distributions are to be computed for:"
## [1] "R1L1Kidney" "R1L2Liver" "R1L3Kidney" "R1L4Liver" "R1L6Liver"
## [6] "R1L7Kidney" "R1L8Liver" "R2L2Kidney" "R2L3Liver" "R2L6Kidney"
explo.plot(mycountsbio, toplot = 1, samples = 1, plottype ="boxplot")
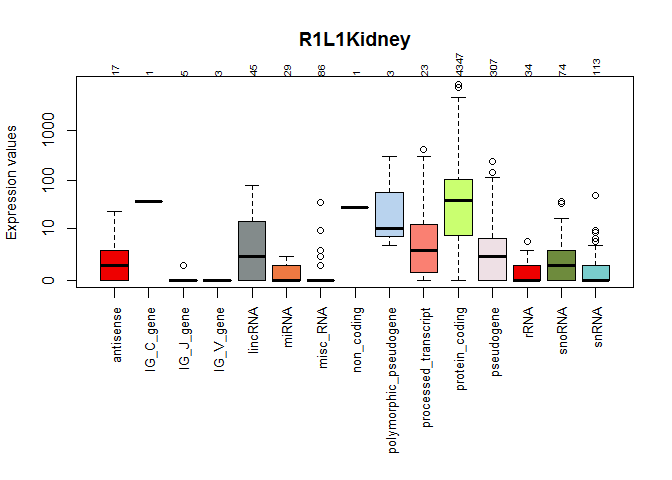
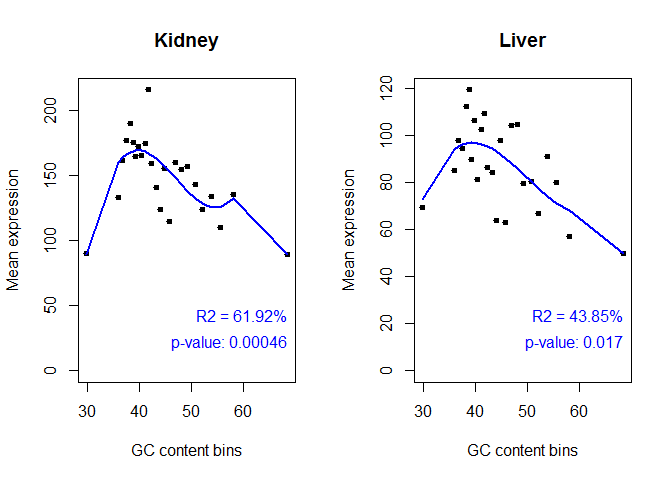
GC Bias
- Used to gauge whether sequencing bias has favored transcripts with low GC content
myGCbias = dat(mydata, factor = "Tissue", type = "GCbias")
## [1] "GC content bias detection is to be computed for:"
## [1] "Kidney" "Liver"
## [1] "Kidney"
##
## Call:
## lm(formula = datos[, i] ~ bx)
##
## Residuals:
## Min 1Q Median 3Q Max
## -38.136 -4.568 0.832 10.406 48.852
##
## Coefficients: (1 not defined because of singularities)
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 89.4686 20.2643 4.415 0.000267 ***
## bx1 0.2447 28.1216 0.009 0.993144
## bx2 177.5119 36.0617 4.922 8.22e-05 ***
## bx3 -68.5025 57.1321 -1.199 0.244534
## bx4 107.6385 64.0141 1.681 0.108220
## bx5 NA NA NA NA
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 20.26 on 20 degrees of freedom
## Multiple R-squared: 0.6192, Adjusted R-squared: 0.543
## F-statistic: 8.13 on 4 and 20 DF, p-value: 0.0004613
##
## [1] "Liver"
##
## Call:
## lm(formula = datos[, i] ~ bx)
##
## Residuals:
## Min 1Q Median 3Q Max
## -29.424 -9.185 0.000 9.384 22.594
##
## Coefficients: (1 not defined because of singularities)
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 49.862 15.000 3.324 0.00338 **
## bx1 22.700 20.816 1.091 0.28845
## bx2 75.753 26.694 2.838 0.01017 *
## bx3 7.595 42.291 0.180 0.85927
## bx4 18.511 47.385 0.391 0.70019
## bx5 NA NA NA NA
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 15 on 20 degrees of freedom
## Multiple R-squared: 0.4385, Adjusted R-squared: 0.3262
## F-statistic: 3.904 on 4 and 20 DF, p-value: 0.01678
explo.plot(myGCbias, samples = NULL, toplot = "global")
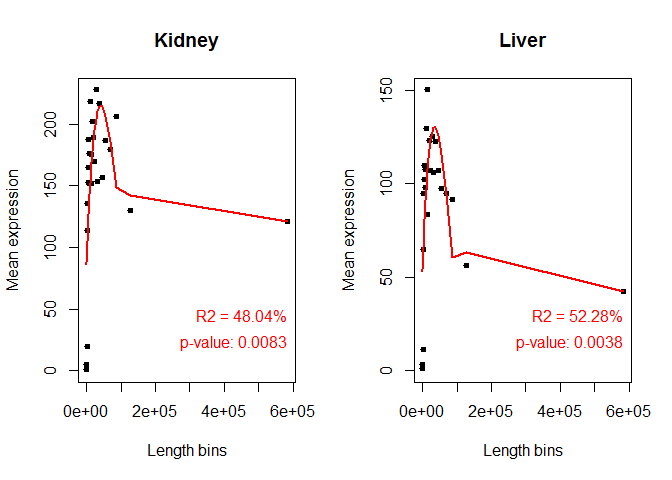
Length Bias
- Used to elucidate if bias is brought about by gene length
mylengthbias = dat(mydata, factor = "Tissue", type = "lengthbias")
## [1] "Length bias detection information is to be computed for:"
## [1] "Kidney" "Liver"
## [1] "Kidney"
##
## Call:
## lm(formula = datos[, i] ~ bx)
##
## Residuals:
## Min 1Q Median 3Q Max
## -85.399 -19.603 3.049 25.848 74.827
##
## Coefficients: (1 not defined because of singularities)
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 121.33 48.63 2.495 0.02148 *
## bx1 -34.97 52.24 -0.670 0.51082
## bx2 269.24 88.41 3.045 0.00639 **
## bx3 -1301.13 719.73 -1.808 0.08570 .
## bx4 6292.42 4655.39 1.352 0.19158
## bx5 NA NA NA NA
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 48.63 on 20 degrees of freedom
## Multiple R-squared: 0.4804, Adjusted R-squared: 0.3764
## F-statistic: 4.622 on 4 and 20 DF, p-value: 0.008332
##
## [1] "Liver"
##
## Call:
## lm(formula = datos[, i] ~ bx)
##
## Residuals:
## Min 1Q Median 3Q Max
## -51.84 -18.19 0.00 21.29 42.30
##
## Coefficients: (1 not defined because of singularities)
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 42.18 29.68 1.421 0.170729
## bx1 10.75 31.88 0.337 0.739524
## bx2 222.14 53.96 4.117 0.000535 ***
## bx3 -1141.68 439.26 -2.599 0.017161 *
## bx4 5758.12 2841.23 2.027 0.056243 .
## bx5 NA NA NA NA
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 29.68 on 20 degrees of freedom
## Multiple R-squared: 0.5228, Adjusted R-squared: 0.4273
## F-statistic: 5.477 on 4 and 20 DF, p-value: 0.003815
explo.plot(mylengthbias, samples = NULL, toplot ="global")
Length Bias, output 2
show(mylengthbias)
## [1] "Kidney"
##
## Call:
## lm(formula = datos[, i] ~ bx)
##
## Residuals:
## Min 1Q Median 3Q Max
## -85.399 -19.603 3.049 25.848 74.827
##
## Coefficients: (1 not defined because of singularities)
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 121.33 48.63 2.495 0.02148 *
## bx1 -34.97 52.24 -0.670 0.51082
## bx2 269.24 88.41 3.045 0.00639 **
## bx3 -1301.13 719.73 -1.808 0.08570 .
## bx4 6292.42 4655.39 1.352 0.19158
## bx5 NA NA NA NA
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 48.63 on 20 degrees of freedom
## Multiple R-squared: 0.4804, Adjusted R-squared: 0.3764
## F-statistic: 4.622 on 4 and 20 DF, p-value: 0.008332
##
## [1] "Liver"
##
## Call:
## lm(formula = datos[, i] ~ bx)
##
## Residuals:
## Min 1Q Median 3Q Max
## -51.84 -18.19 0.00 21.29 42.30
##
## Coefficients: (1 not defined because of singularities)
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 42.18 29.68 1.421 0.170729
## bx1 10.75 31.88 0.337 0.739524
## bx2 222.14 53.96 4.117 0.000535 ***
## bx3 -1141.68 439.26 -2.599 0.017161 *
## bx4 5758.12 2841.23 2.027 0.056243 .
## bx5 NA NA NA NA
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 29.68 on 20 degrees of freedom
## Multiple R-squared: 0.5228, Adjusted R-squared: 0.4273
## F-statistic: 5.477 on 4 and 20 DF, p-value: 0.003815
RNA composition
mycd = dat(mydata, type = "cd", norm = FALSE, refColumn = 1)
## [1] "Reference sample is: R1L1Kidney"
## [1] "Confidence intervals for median of M:"
## 0.28% 99.72% Diagnostic Test
## R1L2Liver "-0.891798603865612" "-0.766824549577716" "FAILED"
## R1L3Kidney "-0.0471703644087826" "-0.0471703644087824" "FAILED"
## R1L4Liver "-0.879624287764932" "-0.7540048872931" "FAILED"
## R1L6Liver "-0.903134087665681" "-0.761454067082924" "FAILED"
## R1L7Kidney "0.0348451027997053" "0.0348451027997056" "FAILED"
## R1L8Liver "-0.904500838561669" "-0.760538326376263" "FAILED"
## R2L2Kidney "-0.0850229820491386" "-0.0468824681954803" "FAILED"
## R2L3Liver "-0.879935949269671" "-0.749483184477486" "FAILED"
## R2L6Kidney "-0.0726984636810064" "-0.0363901813919369" "FAILED"
## [1] "Diagnostic test: FAILED. Normalization is required to correct this bias."
explo.plot(mycd)
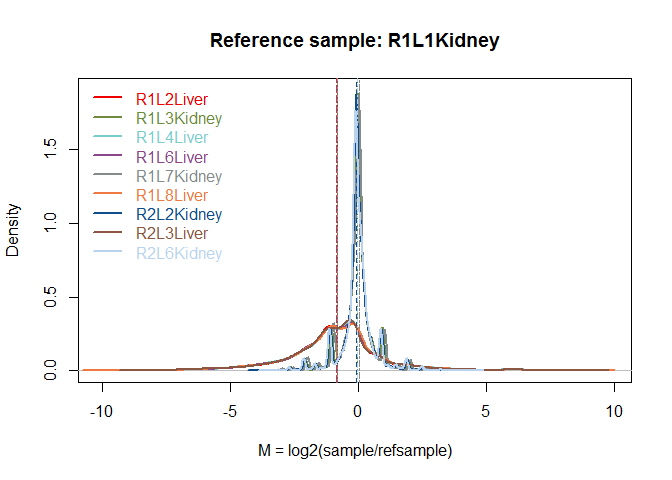
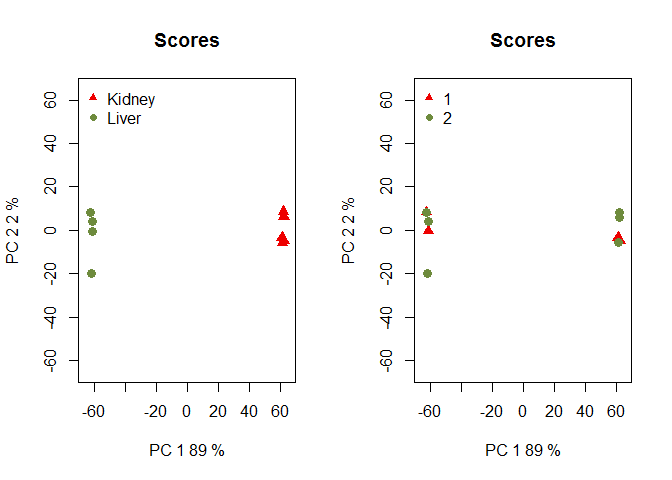
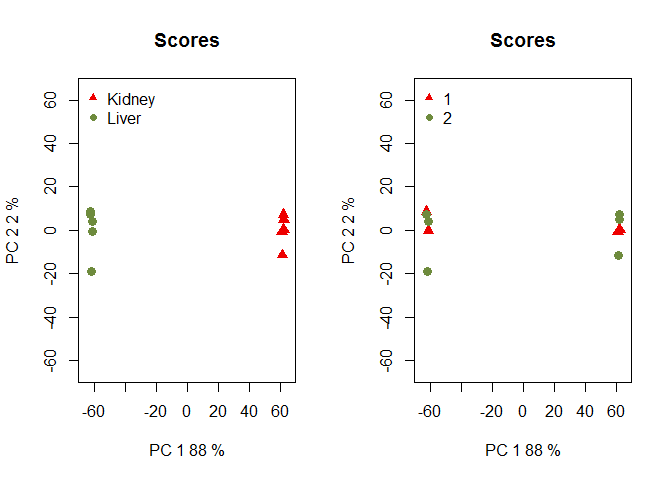
Batch effect
Here, we introduce a batch effect in example, and will later correct for it:
set.seed(123)
mycounts2 = mycounts
mycounts2[, 1:4] = mycounts2[, 1:4]
runif(nrow(mycounts2) * 4, 3, 5)
myfactors = data.frame(myfactors, batch = c(rep(1, 4), rep(2, 6)))
mydata2 = readData(mycounts2, factors = myfactors)
myPCA = dat(mydata2, type = "PCA")
par(mfrow = c(1, 2))
explo.plot(myPCA, factor = "Tissue")
explo.plot(myPCA, factor = "batch")
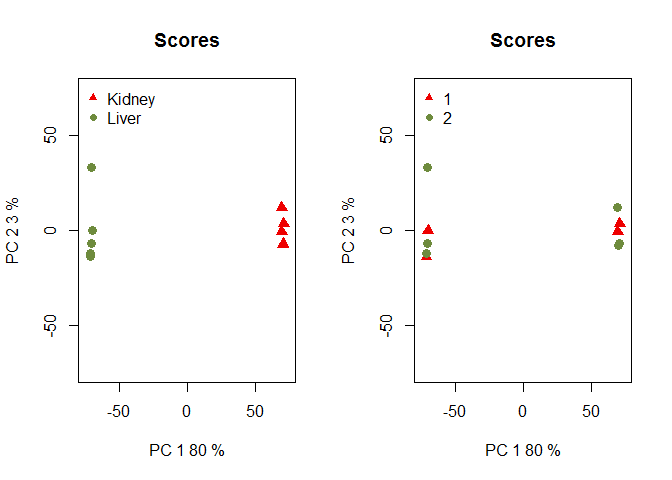
Sequencing Depth
A key question in RNA-Seq:
- Are the number of sequencing reads enough to study our genome and to properly quantify feature expression?
Saturation plot
-
How many features in the genome are being detected with more than a given number of counts, k (k=0 or k=5)
-
How the number of detected features would change with increasing sequencing depth
- For all features or for each biotype
Saturation plot
mysaturation = dat(mydata, k = 0, ndepth = 7, type = "saturation")
explo.plot(mysaturation, toplot = 1, samples = 1:2, yleftlim = NULL,
yrightlim = NULL)
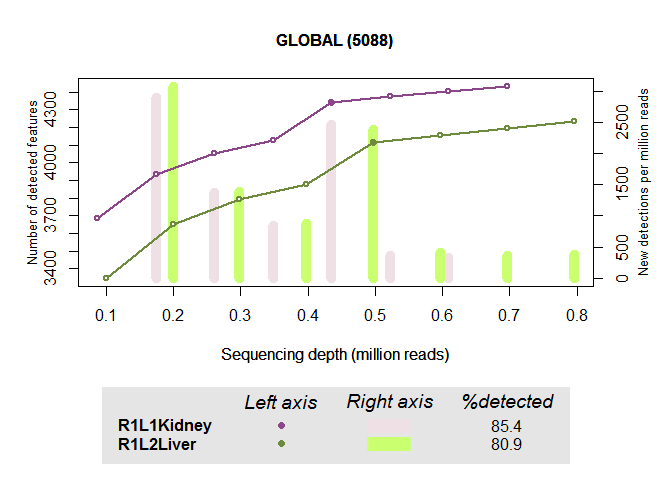
Distribution of protein-coding genes of a specific biotype
explo.plot(mysaturation, toplot="protein_coding", samples=1:4)
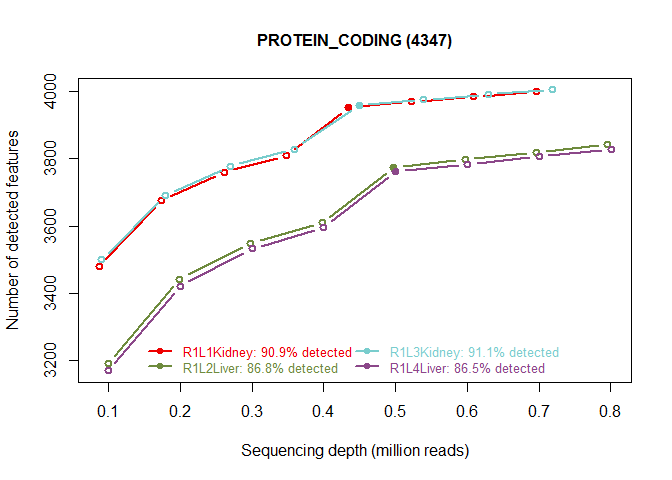
Count distribution for all samples for a certain biotype
explo.plot(mycountsbio, toplot = "protein_coding", samples = NULL,
plottype = "boxplot")

Sensitivity plot
explo.plot(mycountsbio, toplot = 1, samples = NULL, plottype = "barplot")
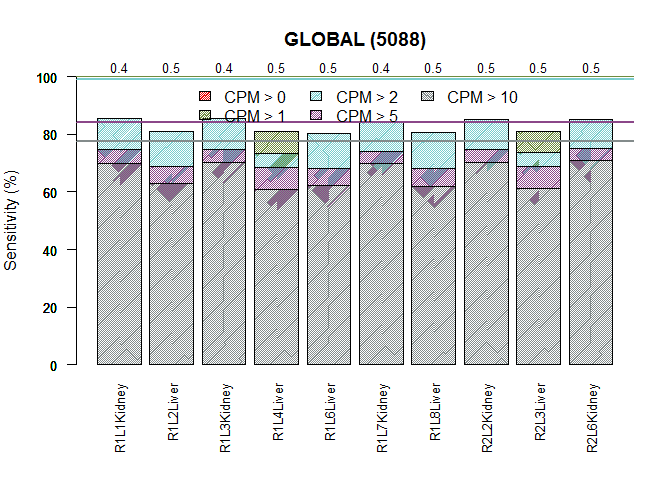
Filtering low counts
- Excluding low counts generally improves differential expression
results
- noise is reduced
- The best procedure to filter low count features is unknown
- NOISeq proposes 3 different methods to filter out features with low
counts:
- CPM Method
- Wilcoxon test
- Proportion test
myfilt = filtered.data(mycounts, factor = myfactors$Tissue, norm = FALSE,
depth = NULL, method = 1, cv.cutoff = 100, cpm = 1)
## Filtering out low count features...
## 4406 features are to be kept for differential expression analysis with filtering method 1
Normalization
- Necessary to analyze between samples and remove sequencing depth bias
- Can normalize in RPKM, Upper Quartile, and TMM
## R1L1Kidney R1L2Liver R1L3Kidney R1L4Liver R1L6Liver
## ENSG00000177757 1.866057 0.8160788 0.0000000 0.0000000 0.8375346
## ENSG00000187634 22.597824 10.8910916 19.1928426 13.6356594 9.5215171
## ENSG00000188976 43.360221 17.6638209 44.2649178 28.9256646 23.9932411
## ENSG00000187961 6.946966 3.2406417 6.7235013 5.2356905 4.5730336
## ENSG00000187583 0.270246 0.0000000 0.2615529 0.2350098 0.0000000
## ENSG00000187642 1.334678 0.0000000 1.6146812 0.0000000 0.5990378
## R1L7Kidney R1L8Liver R2L2Kidney R2L3Liver R2L6Kidney
## ENSG00000177757 1.911677 0.000000 0.879631 0.7854861 2.5794742
## ENSG00000187634 19.370633 14.972295 18.261034 9.7063073 19.9748412
## ENSG00000188976 41.377759 30.302685 39.198749 21.0020365 44.8846064
## ENSG00000187961 6.167891 5.155075 5.239510 7.7978955 6.4019183
## ENSG00000187583 0.830558 0.000000 0.000000 0.4550224 0.7471288
## ENSG00000187642 4.101920 0.309504 2.831161 1.1236213 2.7674127
Normalization Cheat Sheet
-
long = Numeric vector containing the length of the features
-
k, Counts equal to 0 are changed to k in order to avoid indeterminations when applying logarithms. By default, k = 0
-
k = NULL, makes all zero values into a value between 0 and the lowest non-zero value.
-
lc = Correction factor for length normalization. This correction is done by dividing the counts vector by (length/1000)lc. If lc = 0, no length correction is applied. By default, lc = 1 for RPKM and lc = 0 for the other methods.
Batch Effect Correction
- ARSyNseq filters the noise associated to identified or not identified batch effects considering the experimental design and applying Principal Component Analysis (PCA) to the analysis of variance (ANOVA) parameters and residuals.
If the batch for each sample is known:
Batch is unknown
If the batch for each sample is unknown:
NOISeq options
-
Designed to compute differential expression on data:
- With technical replicates (NOISeq-real)
- No replicates at all! (NOISeq-sim)
-
NOISeq-real used when technical replicates are available.
- Summarizes by summing them up
-
NOISeq-real can be used on Biological replicates
- Averages replicates instead of summing.
- Manual recommends use of NOISeqBIO for biological replicates
How NOISeq-real works- Summarized
NOISeq-real estimates the probability distribution for M and D in an empirical way, by computing M and D values for every pair of replicates within the same experimental condition and for every feature. Then, all these values are pooled together to generate the noise distribution*
M = log2-ratio of the two conditions
D = the value of the difference between conditions.
A feature is differentially expressed if its corresponding M and D values are likely to be higher than in noise.
Disclaimer: Biological replicates are necessary if the goal is to make any inference about the population. Deriving differential expression from technical replicates is useful for drawing conclusions about the specific samples being compared in the study but not for extending these conclusions to the whole population
*When using NOISeq-real for biological values are averaged instead of summed up.
NOISeq-sim
NOISeq can simulate technical replicates
-
Assumes that read counts follow a multinomial distribution
-
probabilities for each feature in the multinomial distribution are the probablility of a read to map onto that feature.
-
mapping probability are approximated by using counts in the only sample of the corresponding experimental condition.
NOISeq-real & NOISeq-sim
NOISeq-real
mynoiseq = noiseq(mydata, k= 0.5, norm = "rpkm", factor = "Tissue", pnr = 0.2,
nss = 5, v = 0.02, lc = 1, replicates = "technical")
## [1] "Computing (M,D) values..."
## [1] "Computing probability of differential expression..."
NOISeq-sim
myresults <- noiseq(mydata, factor = "Tissue", k = NULL, norm = "n", pnr = 0.2,
nss = 5, v = 0.02, lc = 1, replicates = "no")
## [1] "Computing (M,D) values..."
## [1] "Computing probability of differential expression..."
NOISeqBIO
Optimized for use on biological replicates (at least 2 per condition).
Too much stats- see page 20 of NOISeq manual
mynoiseqbio = noiseqbio(mydata, k = 0.5, norm = "rpkm", factor = "Tissue",
lc = 1, r = 20, adj = 1.5, plot = FALSE, a0per = 0.9, random.seed = 12345,
filter = 2)
## Computing Z values...
## Filtering out low count features...
## 5088 features are to be kept for differential expression analysis with filtering method 2
## [1] "r = 1"
## [1] "r = 2"
## [1] "r = 3"
## [1] "r = 4"
## [1] "r = 5"
## [1] "r = 6"
## [1] "r = 7"
## [1] "r = 8"
## [1] "r = 9"
## [1] "r = 10"
## [1] "r = 11"
## [1] "r = 12"
## [1] "r = 13"
## [1] "r = 14"
## [1] "r = 15"
## [1] "r = 16"
## [1] "r = 17"
## [1] "r = 18"
## [1] "r = 19"
## [1] "r = 20"
## Computing probability of differential expression...
## p0 = 0.132948760535283
## Probability
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.0000 0.9049 0.9937 0.8680 1.0000 1.0000
Results
To look at the number of differentially expressed genes (DEGs):
Works-cited
##
## Tarazona, S., Garcia-Alcalde, F., Dopazo, J., Ferrer, A., &
## Conesa, A. (2011). Differential expression in RNA-seq: a matter
## of depth. Genome Research, 21(12), 2213-2223.
##
## Tarazona, S., Furio-Tari, P., Turra, D., Di Pietro, A., Nueda,
## M.J., Ferrer, A., & Conesa, A. (2015). Data quality aware
## analysis of differential expression in RNA-seq with NOISeq
## R/Bioc package. Nucleic Acids Research.
Comments